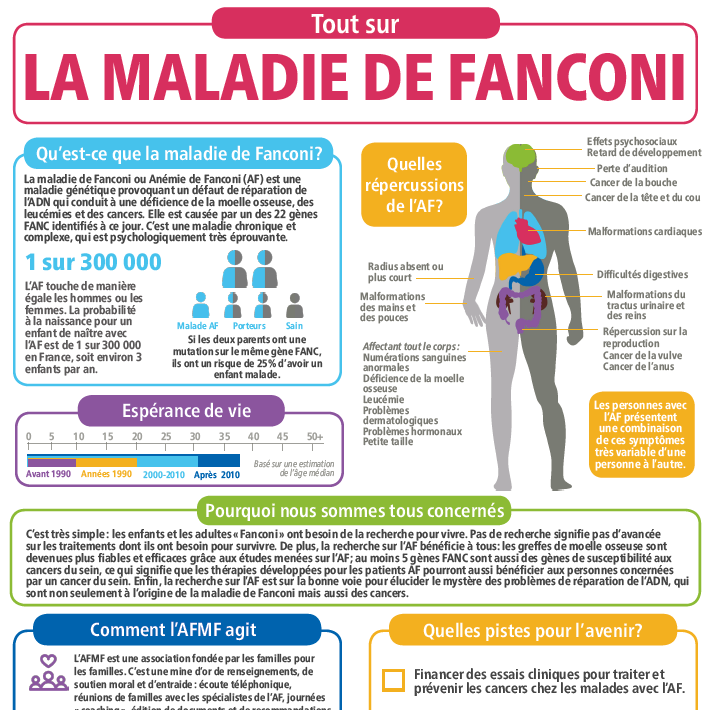
La maladie de Fanconi (ou anémie de Fanconi) est une affection génétique rare de la réparation de l’ADN, avec une prévalence estimée à 1/300.000 naissances, soit environ 300/350 malades en France.
Elle associe des malformations congénitales très variables en gravité et en nombre, une aplasie médullaire progressive, un risque élevé de leucémie aiguë et une prédisposition marquée pour certaines tumeurs solides.
Elle doit son nom au pédiatre suisse Guido Fanconi qui, en 1927, a décrit cette maladie chez des enfants issus de vallées alpines où l’isolement géographique favorisait la consanguinité. Elle survient de façon égale chez les hommes et les femmes ainsi que dans tous les groupes ethniques.
Symptômes et caractéristiques
La maladie de Fanconi est caractérisée par un ensemble de malformations congénitales variables, une insuffisance de la moelle osseuse d’apparition retardée et un risque élevé de leucémie aiguë et de cancers.
Son diagnostic peut être difficile et est rarement fait à un stade précoce. Il est souvent posé en raison des malformations congénitales ou au stade d’apparition de l’insuffisance de la moelle osseuse.
Il existe une très grande hétérogénéité des signes cliniques qui peuvent être très variables d’un individu à un autre et ceci est également vrai au sein d’une même fratrie.
Les principaux signes cliniques
Les premiers signes cliniques sont les malformations congénitales très hétérogènes.
Elles peuvent concerner notamment :
- le squelette : anomalies des pouces ou des radius, microcéphalie (petit périmètre crânien, aspect particulier du visage), petite taille
- les reins et l’appareil urogénital
- le tube digestif (atrésie de l’œsophage, etc.)
- l’appareil auditif
- les yeux
- la peau : tâches café au lait
- les glandes endocrines
- le système nerveux central
- le cœur et les poumons
La petite taille est une des caractéristiques les plus fréquentes : elle débute pendant la vie utérine et les enfants naissent le plus souvent petits. Dans la grande majorité des cas, mais pas pour tous, on observe un retard de croissance modéré mais il peut être plus marqué chez certains patients ou inversement peu marqué. Cette petite taille est souvent due à des déficits hormonaux mais pas toujours.
Il n’y a pas, le plus souvent, de retard du développement psychomoteur ou intellectuel.

Les signes hématologiques
L’âge de survenue d’une atteinte de la moelle osseuse est très variable, y compris au sein d’une même fratrie. Environ ¾ des patients développent une aplasie médullaire modérée ou sévère avant 10 ans, l’âge moyen étant vers 6-7 ans. Cette aplasie est caractérisée par l’insuffisance ou l’arrêt de la production des cellules sanguines dans la moelle osseuse.
Les premiers signes sont des cytopénies (baisse du taux de cellules sanguines) qui débutent le plus souvent par une baisse des plaquettes (thrombopénie) et des globules blancs (neutropénie), associée à une anémie (baisse des globules rouges et macrocytose).
Progressivement s’installe une aplasie médullaire sévère à l’origine d’infections répétées et de besoins en transfusions. Plus de 80% des patients ont évolué vers une aplasie avant l’âge de 20 ans. Une leucémie aiguë, le plus souvent précédée d’un syndrome myélodysplasique, peut venir compliquer cette évolution.
A noter que l’absence de dysfonction de la moelle osseuse n’élimine pas pour autant le diagnostic de maladie de Fanconi.
Les prédispositions aux cancers
Les cancers dans cette maladie sont à la fois plus fréquents que dans la population générale, surviennent à un âge inhabituellement jeune et chez des sujets non exposés aux facteurs de risque classique (tabac, alcool).
Le cancer épidermoïde de la tête et du cou est de loin la tumeur solide la plus fréquente chez les patients avec une maladie de Fanconi. L’incidence de ce cancer chez ces patients est 500 à 700 fois plus élevée que dans la population générale. Il concerne tous les patients, greffés comme non greffés. Ces risques accrus nécessitent une surveillance annuelle systématique dès l’âge de 11-12 ans et de plus en plus fréquente au fur et à mesure que les années passent et en fonction des antécédents.
Les prédispositions aux tumeurs des parties gynécologiques (principalement vulve, vagin et col utérin) sont aussi plus élevées que dans la population générale et impliquent là aussi un suivi systématique dès l’apparition des premières règles.
Pour les personnes non greffées, la survenue d’une leucémie aiguë, précédée d’un syndrome myélodysplasique, est souvent observée. Cette caractéristique de la maladie doit également faire l’objet d’un suivi annuel avec examen cytologique de la moelle osseuse.
D’autres tumeurs sont plus rarement rencontrées et souvent associées à des atteintes des gènes FAND1/BRCA2.



La maladie de Fanconi étant une maladie de la réparation de l’ADN, même si d’un point de vue microscopique les tumeurs/leucémies chez les patients Fanconi sont identiques à celles observées dans la population générale, les traitements diffèrent. En effet, les traitements classiques sont trop agressifs pour les malades Fanconi qui nécessitent des protocoles adaptés à leur fragilité. Il est essentiel dans ces conditions de consulter un spécialiste qui connait bien cette maladie.
Le centre expert de la maladie de Fanconi se trouve à Paris, c’est le Centre de Référence des Aplasies médullaires