La maladie de Fanconi – ou anémie de Fanconi – est une maladie héréditaire rare et évolutive. Elle est marquée par une pancytopénie progressive, c’est à dire une diminution dans le sang des globules rouges (anémie), des globules blancs (neutropénie) et des plaquettes (trombopénie), généralement suivie par une aplasie médullaire qui est une diminution quantitative de la moelle osseuse où sont présentes les cellules souches, éléments précurseurs des cellules du sang. La déficience de la moelle osseuse mène en général à une greffe de moelle osseuse. Il existe également de fortes prédispositions aux hémopathies (maladies du sang) et aux tumeurs solides (cancers).
Cette maladie présente une très grande hétérogénéité phénotypique puisqu’il peut y avoir différentes malformations congénitales associées (squelettiques, cutanées, urogénitales, cardiopulmonaires, digestives, nerveuses centrales etc…).
La maladie de Fanconi est due à des altérations de gènes impliqués dans la réparation de l’ADN ou dans la stabilité des chromosomes. Cette maladie fait ainsi partie des maladies dites « cassantes » de l’ADN.
Ces dix dernières années et grâce aux progrès de la recherche, 23 gènes Fanconi (appelés gènes FANC) ont été découverts.
NB : Ne pas confondre la maladie de Fanconi avec le syndrome de Fanconi (maladie affectant les reins).

Importance du Diagnostic
Le diagnostic est parfois évoqué cliniquement à un stade précoce en raison des malformations congénitales (rein, membre supérieur, cœur…) mais, pour la plupart des patients, il ne l’est qu’au stade de l’insuffisance médullaire, généralement entre l’âge de 4 et 10 ans.
Cependant, l’absence de dysfonction de la moelle osseuse, qui parfois ne se manifeste pas avant l’âge adulte, n’élimine pas pour autant le diagnostic de maladie de Fanconi.
Disposer d’un diagnostic certain est essentiel pour le malade et sa famille. Il permet notamment au médecin de discuter avec le patient et ses proches du pronostic évolutif et de les informer sur les complications possibles. Ceci permet d’avoir le temps de considérer les différentes options thérapeutiques, comme la greffe de moelle osseuse, et de peser leurs avantages et inconvénients respectifs.
Il est également important de tester la fratrie, même s’ils ne présentent aucun symptôme clinique. Cela permet de dépister plus tôt la maladie, quand elle est présente, chez les frères ou sœurs lorsqu’elle ne s’exprime pas encore. Il est également utile de savoir si la fratrie est HLA*-compatible avec la personne atteinte de la maladie. Cela consiste à regarder si les marqueurs de l’immunité de la fratrie sont les mêmes que le malade dans une optique de greffe de cellules souches de la moelle osseuse. Enfin, lorsque les mutations peuvent être identifiées avant une nouvelle grossesse, ceci donne aux parents le temps de considérer les différentes options possibles.
*HLA de l’anglais human leukocyte antigen = CMH complexe majeur d’histocompatibilité = protéines de surfaces d’un organisme permettant de distinguer le « soi » du « non soi ».

Tests Diagnostiques
Test de cassures chromosomiques
Le premier test qui est réalisé quand il y a suspicion de maladie de Fanconi est celui du test des cassures chromosomiques par des molécules cassant l’ADN. C’est un test de cytogénétique où l’aspect physique des chromosomes va être analysé. Ainsi, les cellules sanguines prélevées au cours d’une prise de sang sont traitées par de la Mitomycine C ou par le diépoxybutane, puis étalées sur des lames de verres. Chez des personnes saines, les chromosomes seront d’apparence habituelle, puisque les cellules vont pouvoir se réparer toutes seules. Tandis que chez les personnes atteintes de la maladie de Fanconi, les chromosomes présenteront un nombre important de cassures.
Dans la plupart des cas, ce test suffit à faire le diagnostic, mais il peut néanmoins être d’interprétation difficile ou, de façon rare, apparaitre faussement normal du fait d’une réversion génétique des cellules sanguines appelé mosaïcisme somatique (c’est-à-dire apparition d’une nouvelle mutation génique qui permet de retrouver les fonctions d’un gène après qu’il les a perdues à la suite des premières mutations).
Etude des fibroblastes (cellules de la peau)
Pour la raison citée ci-dessus notamment, une étude complémentaire sur des cellules non sanguines, les fibroblastes cutanés, est souhaitable afin de ne pas passer à côté du diagnostic. Suite à une petite biopsie de peau, les cellules sont étudiées par plusieurs techniques, test de mono-ubiquitination de FANCD2 et test en cytométrie de flux, qui permettent de bien caractériser l’atteinte fonctionnelle sur les fibroblastes, préparant ainsi la recherche des mutations constitutionnelles dans les meilleures conditions.
Un laboratoire unique effectue en France ces analyses spécifiques, le laboratoire d’Hématologie Biologique de l’hôpital Saint-Louis à Paris.
Le processus est déclenché par le médecin prescrivant les examens, qui contacte le laboratoire de St-Louis où est vérifiée la pertinence de l’indication des tests et où est organisé l’envoi des prélèvements.
Identification du gène FANC et caractérisation des mutations
L’ADN extrait des cellules bien caractérisées est ensuite adressé directement du laboratoire de Saint-Louis au laboratoire d’Oncogénétique de l’Institut Curie pour rechercher les mutations (modifications) ou les délétions (absence d’une partie) du gène FANC impliqué.
La première étape est d’identifier quel est le gène FANC impliqué. Pour cela, deux mutations du même gène FANC sont recherchées chez chaque patient, une sur chaque allèle, l’une héritée du père, l’autre de la mère. Il est essentiel de connaître les mutations pour les éventuels futurs diagnostics (nouvelle grossesse). Afin de vérifier la présence de ces mutations, un prélèvement sanguin des parents est aussi envoyé au laboratoire de génétique pour identifier quels sont les allèles paternels et maternels.
Le temps nécessaire à l’identification précise des mutations est variable selon le gène en cause. Il faut compter 4 à 6 mois pour l’ensemble du processus.
Pour en savoir + : lire les chapitres 1 et 2 du guide
Génétique et transmission
Transmission de la maladie
Tous les noyaux des cellules d’un organisme comportent des chromosomes constitués d’ADN qui sont les supports de l’information génétique. Nous avons tous 22 paires de chromosomes (nommés autosomes et numérotés de 1 à 22) et une paire de chromosomes sexuels (XX pour les femmes et XY pour les hommes). Nous héritons d’un chromosome de notre père et l’autre de notre mère. Les chromosomes portent des gènes différents, le génome humain se composant de 20 376 gènes. Chaque gène est caractérisé par une séquence d’ADN particulière et est présent sous deux formes (=allèles), une qui vient de notre père et l’autre de notre mère. Nous pouvons faire une analogie avec les lettres de l’alphabet qui représentent l’ADN et le mot qui représente le gène. Il y a ainsi quatre lettres pour l’ADN qui permet d’écrire un peu plus de 20 000 gènes /mots différents.
La plupart des gènes codent pour une protéine impliquée dans un processus biologique particulier. Nous portons tous des mutations « muettes » au sein de nos gènes qui n’impactent pas la fonction des protéines et qui sont responsables de la diversité de nos caractères.
Lorsqu’une séquence génique porte des variations anormales, que l’on appelle des mutations ou des délétions, il y a alors une altération plus ou moins forte de la fonction du gène et donc de la fonctionnalité de la protéine. Une mutation correspond ainsi à une mauvaise écriture du code du gène tandis qu’une délétion est une partie manquante d’un gène. Si nous reprenons l’analogie avec les mots, supposons que le gène FANCA code pour le mot « plateau », une mutation dans ce gène le transformera en « manteau » tandis qu’une délétion le transformera en « plat ».
Les personnes étant porteuses de mutations/délétions délétères (donc nocives) dans un des 23 gènes Fanconi identifiés à ce jour sont donc atteintes de cette maladie. Ces 23 gènes sont situés sur des chromosomes différents : il y a un gène (FANCB) sur le chromosome sexuel X et les 21 autres sont situés sur les chromosomes autosomes. La transmission de la maladie de Fanconi est ainsi dite autosomique récessive, c’est-à-dire que le gène impliqué est sur un des vingt-deux chromosomes autosomes et qu’il faut avoir hérité de 2 mutations délétères de ce gène pour être malade. Pour le gène FANCB, qui est localisé sur le chromosome X, on parle de transmission liée à l’X.
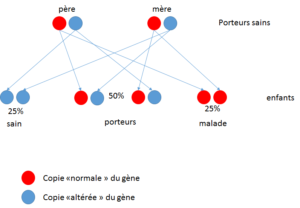
Les personnes ayant une seule copie altérée d’un gène pour une maladie autosomique récessive sont appelées « porteurs sains» (c’est le cas des parents avec un enfant « Fanconi »).
Ces « porteurs » ne développent pas la maladie, mais ils peuvent transmettre une copie du gène altéré à chacun de leurs enfants.
Lorsque les deux parents sont porteurs d’une mutation sur un même gène, il y a trois résultats possibles à chaque grossesse :
- une probabilité de 25% que l’enfant ait deux copies non altérées du gène Fanconi et il est alors indemne de la maladie
- une probabilité de 50% que l’enfant ait une copie du gène altéré FANC et soit donc « porteur » à l’état hétérozygote (un allèle Fanconi, un allèle sain)
- et enfin une probabilité de 25% que l’enfant ait deux copies du gène altéré et qu’il soit alors atteint de la maladie.
Pour en savoir plus lire le chapitre 2 du guide
Conseil génétique
Toutes les personnes atteintes de la maladie de Fanconi, ainsi que leurs familles, devraient être encouragées à bénéficier d’un conseil génétique, soit avec un généticien connaissant bien cette maladie, sa complexité et ses enjeux, soit avec un conseiller en génétique.
Identifier les mutations peut avoir une incidence sur le dépistage du cancer et permet surtout de clarifier les possibilités de diagnostic prénatal et de diagnostic préimplantatoire en vue d’un projet parental.
Les tests génétiques ont de nombreux avantages mais aussi leurs limites. En conséquence, la décision sur l’opportunité de réaliser un test génétique est un choix personnel. Les personnes doivent être informées des enjeux de ces tests pour elles-mêmes et pour les membres de leur famille.
Le conseil génétique est un processus pour aider les personnes à comprendre et à s’adapter aux conséquences médicales, psychologiques et familiales d’une maladie génétique.

Diagnostic prénatal et diagnostic préimplantatoire
Pour les familles qui ont un projet parental, la gravité de cette maladie peut justifier d’avoir recours aux techniques de diagnostic prénatal (DPN) ou de diagnostic préimplantatoire (DPI).
Mais avant tout projet de ce type, il est indispensable de s’assurer qu’un diagnostic génétique précis a été posé pour le membre de la famille atteint, c’est-à-dire qu’il est nécessaire de savoir quel gène est impacté et quelles sont les mutations impliquées.
Le diagnostic prénatal ou DPN
Le diagnostic prénatal est réalisé le plus souvent sur un prélèvement de villosités choriales ou biopsie du trophoblaste à 10 semaines de grossesse ou 12 semaines d’aménorrhée. Cela nécessite d’avoir recours à une échographie de datation à partir de 6 semaines de grossesse.
Les résultats fœtaux (embryon sain ou atteint) sont obtenus sous une dizaine de jours.
Le diagnostic prénatal (DPN) est l’ensemble des pratiques médicales ayant pour but de détecter in utero, chez l’embryon ou le fœtus, une affection grave (anomalie génétique ou malformation congénitale par exemple), afin de donner aux futurs parents le choix d’interrompre ou non la grossesse et de permettre une meilleure prise en charge médicale de la pathologie si la grossesse est poursuivie.
Si les mutations ne sont pas connues, le procédé est le même que précédemment mais le délai est beaucoup plus long puisqu’il faut auparavant rechercher les mutations en cause. En pratique, l’ensemble de cette démarche ne peut généralement être mené à terme dans un délai adéquat pour une grossesse qui est déjà en cours. Il faut donc anticiper largement cette procédure.
Le diagnostic préimplantatoire ou DPI
L’identification précise des mutations peut aussi permettre de réaliser un diagnostic préimplantatoire lors d’une procédure de fécondation in vitro. Après la fécondation in vitro et avant le transfert des embryons dans l’utérus de la mère, une cellule (ou deux) de chaque embryon est prélevée et analysée afin de rechercher l’anomalie génétique en cause. Seuls seront placés dans l’utérus de la maman les embryons indemnes de la maladie génétique.
Le diagnostic préimplantatoire est pratiqué dans seulement cinq centres très spécialisés en France. Cet examen diagnostique est régi par la loi de bioéthique du 6 Août 2004 et ses modalités de fonctionnement sont définies par l’Agence de la Biomédecine.
Le diagnostic préimplantatoire (DPI) est proposé aux couples qui risquent de transmettre à leur enfant une maladie génétique d’une particulière gravité au moment où la démarche de DPI est initiée. L’intérêt de cette technique est de pouvoir réaliser un diagnostic génétique sur un embryon- obtenu par fécondation in vitro – avant qu’il ne soit réimplanté dans l’utérus.